Farmacocinética: Los resultados combinados de dos estudios farmacocinéticos, realizados en 49 adultos portadores de dermatitis atópica indicaron que el tacrolimus ungüento 0.1% se absorbe posterior a su aplicación. Tacrolimus es metabolizado a nivel hepático por el citocromo P-450 3A4 isoenzima vía monodesmetilación, hidroxilación, dedemetilación o una combinación de los caminos de hidroxilación y monodesmetilación, con la mayoría de los metabolitos excretados en bilis. Aproximadamente 99% del tacrolimus circulante se une a la albúmina y al a-ácido glicoproteína.
Las concentraciones pico de tacrolimus circulantes oscilaron desde valores de detección de 0 hasta 20 ng/ml después de una o múltiples dosis del ungüento 0.1%; reportándose concentraciones pico en sangre inferiores a 5 ng/ml en 45 de los 49 pacientes. Los resultados del estudio farmacocinético de tacrolimus ungüento 0.1% en 20 pacientes pediátricos con dermatitis atópica (rangos etáreos de 6-13 años de edad) mostraron concentraciones máximas en sangre inferiores a 1.6 ng/ml en todos los casos. A partir de las concentraciones sanguíneas reportadas, es evidente que el tacrolimus no se acumula sistémicamente por aplicación tópica intermitente durante periodos superiores a un año.
Se desconoce la biodisponibilidad absoluta del tacrolimus tópico. Comparando con los datos históricos del tacrolimus intravenoso, la biodisponibilidad del tacrolimus ungüento 0.01% reportada, en pacientes con dermatitis atópica, es inferior a 0.5 %. En adultos portadores de dermatitis atópica cuya superficie corporal afectada en promedio correspondió a 53%, la exposición del tacrolimus (por ejemplo, área bajo la curva [ABC]) fue aproximadamente 30 veces inferior a la observada en pacientes con trasplante renal y/o hepático, que recibieron tacrolimus vía oral.
Se desconoce cual es el nivel más bajo de tacrolimus tópico circulante en sangre, con el cual se pueden observar efectos sistémicos.
Estudios clínicos: La dermatitis atópica es una enfermedad cutánea inflamatoria crónica asociada a una alteración inmunitaria. Se caracteriza por un exantema pruriginoso, una xerodermia y otra gama de lesiones que varían según la gravedad de la enfermedad: exantema y eritema en las lesiones agudas y engrosamiento cutáneo y liquenificación en las crónicas. Son frecuentes las sobreinfecciones virales, bacterianas o micóticas y en ocasiones son complicaciones graves. La DA puede asociarse a otras enfermedades alérgicas como la rinitis, el asma y la conjuntivitis.
La valoración de la actividad de esta enfermedad es complicada por la morfología variable de las lesiones, lesiones con margen muy pobre y lesiones concurrentes de severidad variable. Además, la piel no involucrada no es completamente normal pero es variablemente seca y puede mostrar histológicamente espongiosis. Existe una falta de estandarización en las medidas de las valoraciones de estas características clínicas para determinar la severidad de la enfermedad. Una revisión reciente evaluó tres escalas de la severidad de DA, las cuales recayeron en la valoración subjetiva de signos y síntomas clínicos y concluyeron que la única escala la cual por su validación, confiabilidad, sensibilidad y aceptabilidad han sido evaluadas fue el índice Scoring of Atopic Dermatitis (SCORAD). El SCORAD es un índice compuesto de observaciones clínicas de la morfología de las lesiones; de la estimación de la extinción del involucramiento usando la regla de los nueves; y la valoración del paciente (o padres) de los síntomas.
Mientras que SCORAD es ampliamente usado como el actual estándar de oro, han aparecido preocupaciones debido a que sólo recae en criterios subjetivos. Además, el método utilizado para estimar el área de superficie corporal (ASC) afectada, la regla de los nueves es también subjetiva. Recientemente otra herramienta de valoración el Eczema Area and Severity Index (EASI), se introdujo para valorar la extensión y la actividad de la DA, pero otra vez este índice recae en parámetros subjetivos.
En el estudio realizado por Sugarman y cols., se diseñó una forma subjetiva de valorar la función del estrato córneo (SC) especialmente, la función de la permeabilidad de la barrera y la hidratación del SC a través de una forma rápida y no invasiva empleando tecnología de bioingeniería; lo cual podría utilizarse para valorar la severidad de la DA. Los valores de la pérdida de agua transepidérmica (PATE), una medición directa de la permeabilidad de la barrera están aumentados en la DA, tanto en la piel involucrada como en la piel no involucrada en relación a la severidad. Los defectos de barrera, en su mayoría, se deben a una disminución en el contenido de ceramida del SC, un lípido clave en la lámina intracelular que forma la permeabilidad de la barrera.
Los esteroides tópicos y los emolientes son el tratamiento habitual de las lesiones agudas de la DA. El uso reiterado de los corticoides tópicos conlleva el riesgo de presentar efectos secundarios locales, como la atrofia cutánea, las estrías y efectos sistémicos. Tacrolimus tópico es el primero de los inmunomoduladores tópicos no esteroideos utilizado como tratamiento de la DA. Su mecanismo de acción se basa en la unión de una proteína citoplásmica, FKBP12. El complejo resultante (FK506) bloquea la actividad de la calcineurina, lo que evita que se active el factor nuclear de activación de los linfocitos T (NFAT).
Esta actividad inhibitoria suprime la transcripción de diferentes genes encargados de la síntesis de numerosas citocinas, como IL-2, por lo que aunque exista una señal de activación de una célula dendrítica, el linfocito T no se activa. Tacrolimus inhibe también la producción de IL-3, IL-4, IL-5, factor estimulante de colonias granulocíticas y macrófagos (GM-CSF) e interferón gamma (INFg), citocinas importantes en la patogenia de la DA. Actúa sobre los síntomas de la DA por medio de sus efectos sobre las células presentadoras de antígeno epidérmicas, los eosinófilos, los mastocitos, los basófilos y los queratinocitos.
González de Olano y cols., realizaron un estudio observacional abierto en las condiciones de práctica clínica para evaluar la eficacia clínica de tracolimus como tratamiento de la DA resistente al tratamiento tradicional. Para ello se seleccionaron pacientes diagnosticados con dermatitis atópica conforme a los criterios de Hanifin y Radka de intensidad moderada-grave a los que se daba seguimiento en el servicio de alergia de nuestro hospital y resistentes al tratamiento habitual empleado hasta entonces: emolientes, antihistamínicos orales y corticoides tópicos. Los criterios de exclusión fueron los siguientes:
Menores de 2 años de edad.
Pacientes que habían recibido vacuna con virus vivos el mes anterior al inicio del tratamiento.
Hipersensibilidad conocida a los macrólidos en general, a tacrolimus o a alguno de sus excipientes.
Se les administró tacrolimus tópico de acuerdo con lapauta recomendada.
Para el seguimiento de los pacientes se diseñó una base de datos en la que se valoraba el índice scorad: la extensión, la intensidad y la gravedad de las lesiones cutáneas antes del inicio del tratamiento con tacrolimus, a las 3 semanas y a las 7 semanas. También se interrogó a los pacientes sobre posibles efectos secundarios.
Finalmente, se procedió a comparar éstos parámetros antes y después del tratamiento con tacrolimus. A los pacientes se les informó las características del estudio antes de incluirlos en él. Se seleccionaron 15 pacientes (7 mujeres y 8 varones) de entre 2 y 34 años (media: 18 años). El tiempo de seguimiento osciló entre 21 y 108 días, con una mediana de 31 y una media de 64.5 días. De los 15 pacientes, 12 (80%) presentaban brotes de manera continua, mientras que 3 (20%) los presentaban con una frecuencia cercana a uno al mes. A lo largo del tiempo que duró el seguimiento, 6 pacientes (40%) presentaron un nuevo brote de la enfermedad, por lo que fue necesario reiniciar el tratamiento conforme al esquema inicial. En un paciente (6.6%), las lesiones remitieron espontáneamente sin necesidad de tratamiento.
Reacciones adversas; 11 de los 15 pacientes (73%) experimentaron prurito y escozor cutáneo en las 24-48 horas siguientes a la administración de tacrolimus que después se resolvieron espontáneamente a pesar de continuar con el tratamiento. Un paciente (9%) refirió rubefacción facial tras el consumo de alcohol que también desapareció en 1 a 2 horas. Se compararon los datos recopilados en la primera y última visitas referentes al índice SCORAD, la extensión de las lesiones, su gravedad e intensidad.
La extensión de las lesiones se registró mediante el porcentaje de superficie corporal afectada conforme a la regla de Wallace.
Para medir la gravedad de las lesiones se evaluaron el eritema, el edema/pápula, la costra/exudación, la excoración, la liquenificación y la sequedad cutánea puntuando cada uno en valores que oscilaban entre 0-3 (lo que conseguía una puntuación máxima posible de 18). Para un mejor análisis final el resultado se estratificó en tres niveles: (0-6), moderado (7-12) e intenso (13-16). Ninguno de los pacientes estudiados se encontraba en éste último nivel por lo que para el análisis estadístico se compararon los niveles leve frente a moderado a grave.
Se procedió de manera similar para evaluar la intensidad de los síntomas; el prurito y el insomnio se puntuaron de 0-10 (con una puntuación máxima posible de 20) y el resultado final se estratificó en cuatro niveles: mínimo (0-5), moderado (6-10), grave (11-15) e insoportable (16-20). Igual que con la variable anterior, en el análisis final se compararon mínimo-moderado frente a grave-insoportable (tabla 2).

Cada variable se analizó conforme a la prueba estadística indicada según sus características. Así, para el estudio del índice SCORAD, donde los valores antes y después de tratamiento con tacrolimus fueron de 41.5 y 18.4, respectivamente, se utilizó la prueba de Wilcoxon, que obtuvo significación estadística con un valor de p = 0.001. De igual manera se procedió para analizar la extensión corporal: el porcentaje de afectación antes y después resultando de 53 y 26,6%, respectivamente. Tras aplicar la prueba de Wilcoxon, el grado de significación estadística obtenido fue de p = 0.003. Para el análisis de la gravedad y la intensidad se compararon los grupos indicados antes; la significación obtenida con la prueba de McNemar fue de p= 0.031 para la gravedad de las lesiones y p = 0.007 para la intensidad (tabla 3).
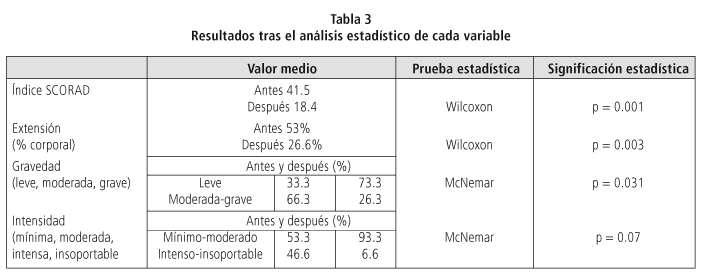
Para finalizar, los autores señalaron que los datos obtenidos en éste estudio piloto, aportan conclusiones similares a las referidas en los ensayos clínicos citados antes. Indican la eficacia de tacrolimus tópico como tratamiento frente a la DA moderada-grave resistente al tratamiento tradicional, tanto en adultos como en niños, con las concentraciones del fármaco recomendadas en cada paciente. Los parámetros globales valorados al inicio y finalización del tratamiento (índice SCORAD) así lo indican con significación estadística.
Se observó además una mayor eficacia en la extensión de las lesiones frente a su gravedad e intensidad.
Si bien los resultados obtenidos en estas dos últimas variables no son estadísticamente significativas, sí nos sirven para valorar la mejoría individual y seguir a cada paciente. La tolerancia de tacrolimus fue reportada como buena; no se han encontrado efectos adversos graves ni diferentes a los referidos hasta ahora. Para el análisis de la eficacia de las variables de nuestro estudio se ha utilizado el índice SCORAD, sistema reconocido en Europa y similar a los mAUC, EASI mEASI, utilizados en los trabajos americanos.
No existe actualmente una alternativa segura para la aplicación tópica de corticosteroides para el control de los episodios agudos de la dermatitis atópica. Aunque los corticosteroides tópicos son generalmente bien tolerados, comúnmente causan atrofia de la piel y con menos frecuencia causan hipopigmentación, infecciones secundarias y acné. La ciclosporina tópica ha sido investigada como un tratamiento alternativo de la dermatitis atópica y otras dermatosis, pero estos estudios han tenido poco éxito debido a la poca penetración del medicamento.
Tacrolimus (FK 506) es un inmunosupresor primario efectivo y bien tolerado usado en el trasplante de órganos. Aunque su mecanismo de acción es similar a la ciclosporina, su peso molecular es menor y su potencia para inhibir la activación de células T es de 10 a 100 veces mayor. Más aún aplicado tópicamente tacrolimus parece penetrar la piel lo suficiente para conseguir un efecto inmunosupresor local. Además inhibe la dermatitis alérgica por contacto inducida experimentalmente y estudios preliminares han sugerido que el medicamento es efectivo en el tratamiento de la dermatitis atópica.
Para valorar la eficacia y seguridad de tacrolimus tópico en pacientes con dermatitis atópica, se realizó un estudio comparativo por Ruzika y cols., a diferentes concentraciones de tacrolimus ungüento 0.03, 0.1, y 0.3%. La primera meta fue una puntación combinada para eritema, edema y prurito. El estudio se llevó a cabo en 16 centros en Europa entre abril 1995 y marzo 1996. Fue fase 2, al azar, doble-ciego, multicéntrico. Una fase de tres semanas de tratamiento durante la cual se aplicó el aceite a un área sintomática definida de 200 a 1000cm2 de piel, fue precedida por una semana de wash out con la subsecuente semana de seguimiento clínico. En éste estudio se incluyeron a pacientes masculinos y femeninos de 13 a 60 años de edad con diagnóstico confirmado de dermatitis atópica de moderada a severa de acuerdo al criterio de Rajka y Langeland. Se excluyeron todos aquellos pacientes que habían recibido tratamientos tópicos y sistémicos para dermatitis atópica dentro de las tres semanas antes de iniciar la fase de wash out. Los criterios para ingresar a la fase de tratamiento fueron un área sintomática de al menos 200 cm2 de piel en tronco o extremidades o ambos y no evidencia de hipersensibilidad a la base aceitosa.
Al inicio de la fase de tratamiento, 200 a 1,000 cm2 de piel afectada fue seleccionada para el tratamiento. El área afectada podía ser no contigua y podía incluir el tronco, extremidades, cara y cuello; sin embargo, 200cm2 de piel afectada tenían que estar en el tronco o extremidades. Se seleccionaron las lesiones con el eritema y edema más acentuado. Se les asignó a los pacientes recibir 0.03, 0.1 o 0.3% de tacrolimus ungüento o solo, el vehículo (el ungüento base) en la base de 1:1:1:1 esquema al azar estratificado por el centro.
La base del ungüento fue una emulsión de aceite conteniendo carbonato de propileno, cera blanca, aceite mineral, parafina y petrolato. Cada semana durante la fase de tratamiento 8 tubos de 10 g fueron administrados a cada paciente; quienes fueron instruidos de aplicarse el ungüento en el área seleccionada dos veces al día, con una separación de 12 horas. Los investigadores, pacientes y monitores clínicos no estaban enterados de la asignación de los tratamientos.
No se permitió tratamiento concomitante que no fuesen emolientes o baño de aceite, se prohibieron otros tratamientos experimentales, tranquilizantes y píldoras para dormir, así como corticosteroides sistémicos, tópicos, o inhalados, antihistamínicos y antimicrobianos.
Se valoraron los pacientes en condiciones basales, el sitio de aplicación se calculó utilizando figuras transparentes con medidas de 100 ó 1,000 cm2 o por la regla de los nueve. La calificación total del cuerpo fue la suma de las calificaciones individuales en una escala de 0 a 3, por eritema, edema, prurito, supuración o encostramiento, excoriación y liquenificación de toda la piel involucrada, sequedad de la piel no involucrada y pérdida de sueño.
El área seleccionada para tratamiento fue valorada en condiciones basales, después de 3 días, una, dos y tres semanas de tratamiento y una semana después de haber completado el tratamiento. El investigador calificó el área seleccionada en una escala de 0 a 3, para la severidad de los signos y síntomas antes mencionados. El paciente calificó el prurito del área seleccionada en una escala análoga la cual se convirtió en valores de 0 a 3 para su análisis. Al final de la fase de tratamiento una valoración en general de la condición del área tratada (síntomas completamente resueltos, marcadamente, moderadamente, ligeramente mejorado, sin cambios o peor) fue realizada por el investigador y el paciente. Todos los investigadores recibieron un manual y una capacitación para la valoración. Todos los eventos adversos fueron registrados en todos los intervalos del estudio, estuviesen relacionados o no con el medicamento.
Las pruebas de laboratorio fueron realizadas en estado basal y después de una dos y tres semanas de tratamiento, incluyeron valoraciones de variables hematológicas, electrólitos séricos, pruebas de funcionamiento renal, hepático, glucosa sérica e IgE sérico total, también, se analizaron por potenciales efectos tóxicos los niveles totales de tacrolimus en sangre en estos intervalos así como 3 días después del inicio del tratamiento.
La principal meta fue el cambio en la calificación 1 (la suma de las calificaciones para eritema, edema y prurito en el área tratada) del estado basal al final del tratamiento, debido a que el edema y el eritema son marcadores confiables de la actividad de la enfermedad. Metas secundarias incluyeron el cambio del estado basal en la calificación 2 (calificación 1 más la suma de las calificaciones por supuración o encostramiento, excoriación y liquenificación de la piel involucrada y sequedad de la piel no involucrada en el área tratada) y la valoración en general por el investigador y paciente del área tratada.
Para las calificaciones 1 y 2 se calcularon los cambios absolutos y porcentuales del estado basal hasta el día 3 y semanas 1,2 y 3 de tratamiento. Se empleó la prueba Jonckheere. Se realizaron análisis por separado para el tronco y las extremidades, la cabeza y el cuello.
Con el fin de probar el primer objetivo para un efecto central, basal y del tratamiento, se calculó un área bajo la curva ABC para la calificación 1 de cada paciente y se realizó un análisis de varianza.
Un total de 250 pacientes fueron monitoreados y 215 fueron asignados al azar a un grupo de tratamiento, dos pacientes fueron excluidos: uno que nunca recibió tratamiento y uno en el que sólo el estado basal estaba disponible, por lo que 213 pacientes fueron incluidos en el análisis de intención de tratamiento: 54 recibieron tacrolimus 0.03%, 54 tacrolimus 0.1%, 51 tacrolimus 0.3% y 54 recibieron sólo el vehículo (grupo control). Todos los grupos se compararon tomando en cuenta las características demográficas y los estados basales.
En los cuatro grupos, el área promedio seleccionada (800 cm2) fue una quinta parte del total del área afectada. Para el tronco, las extremidades, la cara y el cuello, se observó una diferencia significativa entre los grupos tratados en los cambios de la calificación 1 de la línea base hasta el final del tratamiento (p < 0.001). Las medianas de los descensos porcentuales en las calificaciones para el tronco y las extremidades fueron 66.7, 83.3 y 75% para los grupos que recibieron 0.03, 0.1 y 0.3% de tacrolimus respectivamente y 25% para el grupo control. Para la cara y el cuello, los valores fueron 71.4, 83.3, 83.3 y 25% respectivamente en la mejoría de los síntomas tres días después del inicio del tratamiento.
Un análisis de la varianza confirmó un efecto del tratamiento (p < 0.001) y mostró una diferencia significativa entre cada uno de los grupos de tacrolimus y el grupo control (P < 0.001), sin interacción significativa entre el tratamiento, el centro participante y sin diferencias significativas entre los estados basales de cada grupo. Aunque las medianas de los descensos porcentuales en las calificaciones 1 y 2 con el tiempo fueron mayores en los grupos que recibieron 0.1 y 0.3% que el grupo que recibió 0.03%, un análisis de varianza comparativo a la par no mostró diferencias significativas entre los tres grupos.
Los cambios entre el estado basal y el final del tratamiento en la calificación 2 difirieron significativamente entre los cuatro grupos (p < 0.001). Para el tronco y las extremidades, las medianas de los descensos de la línea base al final del tratamiento fueron 61.5, 71.4 y 70% para los tres grupos 0.03, 0.1 y 0.3% respectivamente y 21.8% para el grupo control. Los valores para la cara y cuello fueron 70.6, 75, 77.8 y 27.3% respectivamente, llegando a ser similares con el tiempo estos porcentajes a los de la calificación 1.
De acuerdo a la valoración del investigador una gran proporción de pacientes en los grupos de tacrolimus resolvieron o mejoraron marcadamente su sintomatología a diferencia del grupo vehículo (p < 0.001). Los resultados fueron similares en la valoración de los pacientes (p < 0.001). No hubo diferencias significativas entre los grupos tratados en la incidencia de eventos adversos. Treinta y dos de los 54 pacientes que recibieron 0.03%, 33 de 54 con 0.1%, 32 de los 51 con 0.3% de tacrolimus y 23 de 54 que recibieron sólo vehículo presentaron por lo menos un evento adverso. La sensación de quemazón en el sitio de aplicación fue el único evento con una incidencia significativamente alta en los grupos de tacrolimus a diferencia del grupo control (p < 0.001). Los otros eventos adversos más frecuentemente reportados en el sitio de aplicación fueron prurito (7, 2, 7, y 4 pacientes respectivamente) y eritema(3, 6, 6, y 3 pacientes respectivamente. El evento más frecuente que involucró áreas no tratadas fue una exacerbación de la dermatitis atópica, la cual fue reportada por 4, 4, 2 y 7 pacientes respectivamente.
En los grupos de tacrolimus, todos los eventos adversos que obligaron a la salida del estudio ocurrieron en el sitio de aplicación; 1 paciente con foliculitis en el grupo tratado con 0.03%, 3 con quemazón y 1 con prurito en el grupo tratado con 0.1%, 2 con quemazón y otro con una sospecha de enfermedad viral en la piel en el grupo tratado con 0.3%. En el grupo control 2 pacientes con eventos adversos en el sitio de aplicación (quemazón y prurito respectivamente) fueron retirados, así como 3 pacientes con eventos adversos ajenos al sitio de aplicación (exacerbación de la dermatitis atópica).
La concentración total sanguínea de tacrolimus fue muy baja durante el estudio. Una larga proporción de pacientes tuvo valores inferiores a la prueba de detección (0.05 ng/ml) en todos los intervalos y sólo un pequeño número de pacientes tuvo concentraciones mayores a 1 ng/ml. La mediana de los valores para las pruebas clínicas de laboratorio fue similar en los 4 grupos y no cambiaron apreciablemente durante el estudio. De manera similar no hubo aparentes diferencias en la frecuencia de los cambios individuales en ninguna variable.
En este estudio controlado cada una de las 3 presentaciones en ungüento de tacrolimus fue más efectiva que el vehículo solo. No hubo diferencias estadísticamente significativas entre los tratamientos activos, aunque hubo una tendencia hacia la ventaja de tacrolimus al 0.1% sobre 0.03%. En pacientes con psoriasis que recibieron tacrolimus oral se reportó una aparente toxicidad en concentraciones de 10 ng/ml en un periodo de 9 semanas. En receptores de trasplantes de órgano, los niveles se mantuvieron por debajo de 20 ng/ml para reducir el riesgo de toxicidad.
La exposición sistémica fue baja en este estudio y el descenso de las concentraciones con el tiempo fue disminuyendo; lo que es consistente con otro estudio reciente, es que la absorción de tacrolimus en el torrente sanguíneo disminuyó al resolverse los cuadros en los pacientes con dermatitis atópica sin absorción a través de la piel de pacientes sanos. La sensación de quemazón en el sitio de aplicación parece haber sido el único efecto adverso relacionado con el medicamento. Siendo que una larga proporción de pacientes que recibieron sólo el vehículo también reportaron este evento, la quemazón pudo haber sido causada parte por el vehículo.
Para pacientes con lesiones en la cara, cuello o sitios de flexión, donde la piel es particularmente delgada, proveer una alternativa a los corticosteroides tópicos que no produzca atrofia es crucial. A una concentración que tuvo una eficacia similar al propionato de clobetasol 0.13% (un corticosteroide de alta potencia que causa atrofia) en el manejo de una reacción inflamatoria cutánea en la piel de cerdo, tacrolimus tópico no causó atrofia. El tratamiento con ciclosporina oral en pacientes con dermatitis atópica causa incremento en las concentraciones séricas de urea, creatinina y bilirrubina. La ausencia de nefrotoxicidad puede ser una ventaja importante para el tratamiento tópico con tacrolimus. El tratamiento a corto plazo se recomienda con ciclosporina debido a la nefrotoxicidad, y muchos de los pacientes presentan una recaída dentro de las 6 semanas después de dejar el medicamento.
Se concluyó que tacrolimus ungüento es efectivo en el tratamiento de la dermatitis atópica y que el único evento adverso relacionado con el medicamento parece ser la sensación de quemazón en el sitio de aplicación. En circunstancias especiales del estudio en donde tacrolimus fue aplicado en un área restringida en un periodo de 3 semanas, el tratamiento tuvo un perfil de seguridad aceptable. Es importante obtener datos en la seguridad de tacrolimus administrado en una mayor área de la piel por un periodo más largo. Actualmente publicaciones realizadas por la FDA indican que el uso a largo plazo de tacrolimus tópico se desconoce.
Nakagagua y cols; reportaron la seguridad y eficacia de tacrolimus tópico en la DA. En este estudio, todos los pacientes mejoraron la severidad del padecimiento para el día 21. Además, los niveles séricos registrados más altos de tacrolimus fueron 0.9 ng/ml que son significativamente menores que los niveles (5-20 ng/ml) obtenidos durante la terapia sistémica. Desde entonces, múltiples estudios controlados y no controlados han confirmado la eficacia de tacrolimus tópico para la DA. Por ejemplo, Alaiti, et al demostró la eficacia de tacrolimus aceite a 0.3% aplicado dos veces al día durante 8 días en 31 adultos y 8 niños con DA de moderada a severa.
En este estudio, no se observó acumulación sistémica de tacrolimus; 95% de los pacientes presentaron mejoría notable. No se observó ningún cambio relacionado con medicamentos en el perfil de laboratorio.
En un estudio multicéntrico, aleatorizado, doble-ciego, que comparó la eficacia de tacrolimus con las siguientes concentraciones: 0.03, 0.1 y 0.3% resultó que en 213 pacientes con dermatitis atópica de moderada a severa, se observó mejoría estadísticamente significativa en todos los grupos tratados comparado contra placebo. Sin embargo, diferencias entre los tres grupos de tratamiento no fueron estadísticamente significativas. En concordancia con observaciones previas, el único evento adverso notable durante la terapia fue irritación en el sitio tratado. Además, a lo largo de la investigación la mayoría de los pacientes mantuvo concentraciones séricas debajo de 0.25 ng/ml.
De cualquier manera, se debe hacer notar que en un estudio diseñado para detectar los niveles de concentración sérica de tacrolimus durante el tratamiento tópico de DA, se registró una concentración sérica de 20 ng/ml en un paciente eritrodérmico seis horas después de haber recibido una aplicación de 10 g de aceite con una dosis de tacrolimus y 1 mg/gm.
Aunque las concentraciones séricas disminuyeron a 2.9 ng/ml a las 72 horas y no se observaron efectos adversos sistémicos, este hallazgo apoya la utilidad del monitoreo por laboratorio durante la terapia tópica de tacrolimus especialmente en pacientes con un extenso daño en la piel.
Otros reportes mencionan que para evaluar tacrolimus ungüento en el tratamiento de pacientes con dermatitis atópica moderada a severa, se realizaron 3 estudios fase III aleatorizados, multicéntricos, doble-ciego y controlados por vehículo. En el estudio pediátrico, se incluyeron 351 pacientes con rangos etáreos de 2-15 años de edad, mientras que los otros dos estudios realizados en adultos, incluyeron un total de 632 pacientes con rangos etáreos de 15-79 años de edad. El 55 % de los pacientes correspondieron a mujeres y 27 % eran de raza negra.
Al inicio del estudio, 58 % de los pacientes eran portadores de una dermatitis atópica grave, con un promedio de superficie corporal afectada de 46 %. En más de 80 % de los pacientes, los sitios más afectados correspondieron a la cara y el cuello. En estos estudios, los pacientes utilizaron tacrolimus ungüento a dos concentraciones diferentes: 0.03 y 0.1 %, mientras que el grupo control fue tratado con el vehículo del ungüento dos veces al día; aplicándolo sobre la superficie corporal afectada durante un periodo de 12 semanas.
El grupo pediátrico tratado con tacrolimus ungüento 0.03 % comparado con el grupo control mostró un porcentaje estadísticamente significativo (p < 0.001) de pacientes con una mejoría de 90%; con base en la evaluación global de la respuesta clínica realizada por el médico (el objetivo de eficacia primaria) sin embargo, tacrolimus ungüento 0.1 % no reportó mayor eficiencia comparado con el tacrolimus ungüento 0.03 %.
En los dos estudios realizados con adultos, un porcentaje significativamente mayor (p < 0.001) de pacientes tratados con tacrolimus ungüento al 0.03 % comparado con el grupo control, mostró una mejoría de 90% de acuerdo a la evaluación global de la respuesta clínica realizada por el médico (el objetivo de eficacia primaria predefinido); sin embargo, no existieron evidencias suficientes que demostraran que tacrolimus ungüento 0.1% brindara mayor eficacia que Tacrolimus ungüento 0.03%.
La diferencia en la eficacia entre tacrolimus ungüento 0.1% y tacrolimus ungüento 0.03% se demostró en las fases iniciales del tratamiento de pacientes con dermatitis atópica severa con una extensa superficie corporal afectada así como en adultos de raza negra. El siguiente cuadro muestra los niveles de respuesta en cada grupo de tratamiento según los grupos etáreos.
Se reportó una diferencia estadísticamente significativa entre dos pacientes adultos tratados con tacrolimus ungüento 0.1%, quienes presentaron 90% de mejoría durante la semana 1 de tratamiento y aquellos pacientes tratados con tacrolimus 0.03% en la semana 3 de tratamiento. Asimismo, se demostró una diferencia estadísticamente significativa en el porcentaje de pacientes pediátricos tratados con tacrolimus ungüento 0.03% con 90% de mejoría en la semana 2 de tratamiento.
El 35% de los pacientes adultos tratados con tacrolimus ungüento 0.03% y 41% de los tratados con tacrolimus ungüento 0.1 %, que habían alcanzado una mejoría ³ 90% al final del tratamiento, registraron una regresión en el nivel de mejoría durante las 2 semanas posteriores al finalizar el tratamiento. El 54 % de los pacientes pediátricos tratados con Tacrolimus ungüento 0.03% que habían alcanzado una mejoría ³ 90%, registraron una regresión en el nivel de mejoría durante las 2 semanas posteriores al finalizar el tratamiento.
Al no haberse realizado un seguimiento mas allá de las 2 semanas posteriores al finalizar el tratamiento; se desconoce si otros pacientes registraron alguna regresión en la mejoría clínica.
En ambos grupos de pacientes adultos tratados con tacrolimus ungüento 0.1% y en el grupo de pacientes pediátricos tratados con tacrolimus ungüento 0.03%, se observó una mejoría estadísticamente significativa comparada con el grupo control (p ³ 0.001) tanto en el objetivo de eficacia secundaria, en el porcentaje de la superficie corporal afectada, en la evaluación del paciente relativa al prurito, en el eritema, en el edema, en la excoriación, en la infiltración, en la descamación y en la liquenificación. La evolución en el tiempo de la mejoría del resto de las variables de eficacia secundaria, fue similar a la observada en el eritema; registrándose una mejoría más lenta en la liquenificación.
En otro estudio de eficacia y seguridad terapéutica, pacientes adultos y pacientes pediátricos fueron tratados con tacrolimus ungüento 0.03% presentando una mejoría estadísticamente significativa comparada con el grupo control (p ³ 0.001) en el objetivo de eficacia secundaria, en el porcentaje de la superficie corporal afectada, en la evaluación del paciente relativa al prurito, en el eritema, en el edema, en la excoriación, en la infiltración, en la descamación y en la liquenificación.
Con el objetivo de demostrar la seguridad prolongada de tacrolimus ungüento 0.1%, se realizó un estudio en el cual se incluyó un total de 571 pacientes adultos y pediátricos quienes utilizaron tacrolimus ungüento 0.1% en estudios durante un periodo de 6 y 12 meses. En el estudio con adultos, se evaluaron 246 pacientes durante un periodo de 6 meses y 68 pacientes, durante un periodo de 12 meses. En otro estudio se evaluaron 219 pacientes pediátricos durante un periodo de 6 meses y 180 pacientes adultos durante 12 meses. En promedio, todos los pacientes estudiados recibieron tratamiento con tacrolimus ungüento 0.1% durante el 87% de los días que duró el estudio.




