Grupo farmacoterapéutico: Inhibidor no esteroide de la aromatasa (inhibidor de la biosíntesis de estrógenos); antineoplásico (código ATC: L02B G04).
Propiedades farmacocinéticas:
Absorción: Letrozol se absorbe rápida y completamente del tracto gastrointestinal (biodisponibilidad absoluta media: 99.9%). Los alimentos disminuyen un poco la velocidad de absorción (media de Tmáx: 1 hora en ayuno vs 2 horas con alimentos; y media de Cmáx: 129 ± 20.3 nmol/l en ayuno vs 98.7 ± 18.6 nmol/l con alimentos), pero no cambia el grado de absorción (ABC).
El efecto menor sobre la velocidad de absorción no se considera clínicamente relevante y, por lo tanto, letrozol puede administrarse sin tomar en cuenta el horario de alimentos.
Distribución: La unión de letrozol a proteínas plasmáticas es de aproximadamente 60%, principalmente a la albúmina (55%). La concentración de letrozol en los eritrocitos es de alrededor del 80% de la plasmática. Después de la administración de 2.5 mg de letrozol marcado con C14, aproximadamente 82% de la radiactividad en el plasma fue encontrado como compuesto no modificado.
La exposición sistémica a los metabolitos, por lo tanto, es escasa. Letrozol se distribuye rápida y extensamente en los tejidos. Su volumen de distribución aparente en estado estacionario es alrededor de 1.87 ± 0.47 l/kg.
Metabolismo y eliminación: La depuración metabólica hasta un metabolito carbinol farmacológicamente inactivo es la vía de eliminación principal de letrozol (CLm = 2.1 l/h), pero es relativamente lenta cuando se compara con el flujo sanguíneo hepático (alrededor de 90 l/h). Se descubrió que las isoenzimas del citocromo P-450, 3A4 y 2A6, son capaces de convertir letrozol en este metabolito. La formación de metabolitos menores no identificados y la excreción renal y fecal directas tienen sólo un papel menor en la eliminación global de letrozol.
En las dos semanas siguientes a la administración de 2.5 mg de letrozol marcado con C14 en voluntarias posmenopáusicas sanas, 88.2 ± 7.6% de la radiactividad fue recuperada en la orina y 3.8 ± 0.9% en las heces. Por lo menos 75% de la radiactividad recuperada en la orina en un lapso hasta de 216 horas (84.7 ± 7.8% de la dosis) fue atribuida al glucurónido del metabolito carbinol, alrededor de 9% a dos metabolitos no identificados y 6% a letrozol no modificado.
La vida media aparente de eliminación terminal en el plasma es de aproximadamente 2 días. Después de la administración diaria de 2.5 mg, los niveles en estado estable se alcanzan en 2 a 6 semanas.
Las concentraciones plasmáticas en estado estable son aproximadamente 7 veces más elevadas que las concentraciones medidas después de una dosis única de 2.5 mg, mientras que son 1.5 a 2 veces más elevadas que los valores de estado estable pronosticados a partir de las concentraciones medidas después de una dosis única, indicando una ligera no-linealidad en la farmacocinética de letrozol con la administración diaria de 2.5 mg. Como los niveles de estado estable se mantienen con el tiempo, puede concluirse que no ocurre una acumulación continua de letrozol. La edad no tuvo ningún efecto en la farmacocinética de letrozol.
Poblaciones especiales: En un estudio en el que participaron voluntarios con diversos grados de función renal (depuración de creatinina en 24 horas de 9-116 ml/min), no se encontró ningún efecto sobre la farmacocinética de letrozol después de una dosis única de 2.5 mg.
En un estudio similar, en el que participaron sujetos con diversos grados de función hepática, las medias de los valores del ABC de los voluntarios con alteración hepática moderada (calificación B de Child-Pugh) fueron 37% más altos que en sujetos normales, pero aún dentro del rango observado en sujetos sin alteración de la función.
En un estudio que comparó la farmacocinética de letrozol después de una dosis oral única en ocho sujetos con cirrosis hepática y disfunción hepática severa (calificación C de Child-Pugh), y en voluntarios sanos (n = 8), el ABC y t½ aumentaron en 95 y 187%, respectivamente.
Por lo tanto, se espera que las pacientes con cáncer mamario y disfunción hepática severa sean expuestas a niveles más altos de letrozol que aquellas pacientes sin disfunción hepática severa.
Sin embargo, ya que en pacientes que recibieron dosis de 5 ó 10 mg/día no se observó un aumento en toxicidad, una disminución de dosis en pacientes con disfunción hepática severa parece no ser requerida, aunque dichas pacientes deben mantenerse bajo una supervisión muy estrecha.
Además, en dos estudios bien controlados donde participaron 359 pacientes con cáncer de mama avanzado, no se observó efecto alguno debido a la disfunción renal (depuración calculada de creatinina: 20-50 ml/min) o hepática en la concentración de letrozol.
Efectos farmacodinámicos: La eliminación de los efectos estimulantes mediados por los estrógenos es un requisito previo para lograr la remisión del tumor cuando el crecimiento del tejido tumoral depende de la presencia de estrógenos. En las mujeres posmenopáusicas, los estrógenos se producen fundamentalmente por acción de la enzima aromatasa, que convierte los andrógenos suprarrenales -principalmente la androstenodiona y la testosterona- en estrona (E1) y estradiol (E2).
La inhibición de la biosíntesis de estrógenos en los tejidos periféricos y en el propio tejido canceroso puede lograrse, pues, mediante la inhibición específica de la enzima aromatasa.
El letrozol es un inhibidor no esteroide de la aromatasa. Inhibe la enzima aromatasa uniéndose de forma competitiva al grupo hemo de la subunidad citocromo P-450 de la enzima, lo cual conduce a una disminución de la biosíntesis de estrógenos en todos los tejidos.
En las mujeres posmenopáusicas sanas, la administración de dosis únicas de 0.1, 0.5 y 2.5 mg de letrozol reduce las concentraciones plasmáticas de la estrona y del estradiol en 75-78% y 78%, respectivamente, respecto a los valores iniciales. La depresión máxima se observa en un plazo de 48 a 78 horas.
En las mujeres posmenopáusicas con cáncer de mama avanzado, la administración de dosis diarias de 0.1 a 5 mg reduce la concentración plasmática del estradiol, la estrona y el sulfato de estrona de 75 a 95%, respecto a los valores iniciales, en todas las pacientes tratadas. Con dosis iguales o superiores a los 0.5 mg, por lo general, las cifras de estrona y de sulfato de estrona son inferiores al límite de detección analítica, lo cual indica que estas dosis logran una mayor depresión estrogénica. En todas estas pacientes la depresión estrogénica se mantuvo a lo largo de todo el tratamiento.
El letrozol inhibe de forma específica la actividad de la aromatasa. No se ha observado alteración de la esteroidogénesis suprarrenal. Tampoco se han apreciado cambios clínicamente significativos en las concentraciones plasmáticas de cortisol, aldosterona, 11-desoxicortisol, 17-hidroxiprogesterona y ACTH, ni en la actividad de la renina plasmática en las pacientes posmenopáusicas tratadas con dosis diarias de letrozol variables entre 0.1 y 5 mg. La prueba de estimulación con ACTH, practicada al cabo de 6 y 12 semanas de tratamiento con dosis diarias de 0.1, 0.25, 0.5, 1, 2.5 y 5 mg, no indicó pérdida alguna de la producción de aldosterona ni de cortisol. En consecuencia, no se requieren suplementos de glucocorticoides ni tampoco de mineralocorticoides.
Las concentraciones plasmáticas de los andrógenos (androstenodiona y testosterona) no experimentan cambios en las mujeres posmenopáusicas sanas tratadas con dosis únicas de 0.1, 0.5 y 2.5 mg de letrozol, y tampoco lo hacen las concentraciones plasmáticas de la androstenodiona en las pacientes posmenopáusicas que reciben dosis diarias de 0.1 a 5 mg, lo cual indica que el bloqueo de la biosíntesis de estrógenos no redunda en una acumulación de precursores androgénicos. El letrozol no modifica las concentraciones plasmáticas de LH y FSH en las pacientes, ni tampoco la función tiroidea, a juzgar por la captación de T3, T4 y TSH.
Tratamiento adyuvante: En un estudio multicéntrico con diseño doble-ciego, más de 8,000 mujeres posmenopáusicas que fueron objeto de una resección quirúrgica del cáncer de mama incipiente con receptores hormonales positivos fueron asignadas de forma aleatoria a uno de los siguientes grupos:
A. Tamoxifeno durante 5 años.
B. FEMARA® durante 5 años.
C. Tamoxifeno durante 2 años y luego FEMARA® durante 3 años.
D. FEMARA® durante 2 años y luego tamoxifeno durante 3 años.
Los resultados de los grupos de monoterapia (grupos A y B), así como los resultados obtenidos 30 días después del cambio de tratamiento en los dos grupos de tratamiento secuencial (grupos C y D) se señalan en la tabla 2. El análisis de la monoterapia frente a los tratamientos endocrinos secuenciales se llevará a cabo cuando se haya reunido la cantidad suficiente de eventos.
Se observó a las pacientes durante una mediana de 26 meses, el 76% de las pacientes durante más de dos años y el 16% (1,252) por un plazo de 5 años o más.
El criterio de valoración principal del ensayo fue la supervivencia libre enfermedad (SLE), que se determinó como el tiempo transcurrido desde la aleatorización hasta que se producía el primer acontecimiento de recidiva locorregional o a distancia (metástasis) de la enfermedad primaria, el desarrollo de cáncer de mama contralateral invasivo, la aparición de un segundo tumor primario no mamario o la muerte por cualquier causa. FEMARA® redujo el riesgo de recidiva en 19% en comparación con el tamoxifeno (RRI, razón de riesgos instantáneos o hazard ratio de 0.81; p = 0.003). Las tasas de SLE al cabo de cinco años fueron del 84.0% con FEMARA® y del 81.4% con el tamoxifeno. La mejora de la SLE con FEMARA® se apreció ya a los 12 meses y se mantuvo después de cinco años. FEMARA® también redujo de forma significativa el riesgo de recidiva en comparación con el tamoxifeno con independencia de si se administraba quimioterapia adyuvante previa (RRI de 0.72; p = 0.0018) o no se administraba dicha terapia (RRI de 0.84; p = 0.004).
En cuanto al criterio de valoración secundario, la supervivencia general, se registraron 358 muertes en total (166 con FEMARA® y 192 con el tamoxifeno). No se apreció ninguna diferencia significativa de supervivencia general entre los tratamientos (RRI: 0.86; P = 0.15). Un criterio de valoración sustituto de la supervivencia general, la supervivencia sin enfermedad a distancia (metástasis), difirió significativamente tanto globalmente (RRI de 0.73; p = 0.001) como en subcategorías de estratificación previamente especificadas. FEMARA® disminuyó significativamente el riesgo de fallo sistémico en 17% en comparación con el tamoxifeno (RRI de 0.83; p = 0.02), y también redujo el riesgo de cáncer de mama contra lateral invasivo en casi 40%, pero debido a la potencia estadística relativamente reducida de tan pocos sucesos, este resultado no fue estadísticamente significativo. Las pacientes que recibieron FEMARA® presentaron un menor número de neoplasias malignas secundarias que las que recibieron el tamoxifeno (1.9% frente al 2.4%). En particular, la incidencia de cáncer de endometrio con FEMARA® fue inferior a la del tamoxifeno (0.2% frente a 0.4%).
Los resultados se resumen en las tablas 2 y 3.

Tratamiento adyuvante extendido: En un estudio multicéntrico, con doble enmascaramiento, aleatorización y control con placebo realizado en más de 5,100 pacientes posmenopáusicas con cáncer de mama primario y presencia confirmada o desconocida de receptores, las pacientes que no presentaban enfermedad tras completar el tratamiento adyuvante con tamoxifeno (de 4.5 a 6 años) fueron asignadas de forma aleatoria al grupo de FEMARA® o del placebo.
El análisis final al cabo de un periodo de seguimiento de una mediana de 28 meses de duración (el 25% de las pacientes fueron objeto de un seguimiento de hasta 38 meses) indicó que FEMARA® reducía el riesgo de recidiva en 42% en comparación con el placebo (rri: 0.58; P = 0.00003).
La sensibilidad del análisis confirmó la solidez de los datos.
El beneficio estadísticamente significativo con respecto a la supervivencia sin enfermedad en favor del letrozol se observó con independencia del estado ganglionar (ganglios no afectados, RRI: 0.48, P = 0.002; ganglios afectados, RRI: 0.61, p = 0.002).
Con respecto al criterio de valoración secundario, la supervivencia general (SG), hubo un total de 113 fallecimientos (51 en el grupo de FEMARA®, 62 en el del pacebo). En general, no se apreció ninguna diferencia significativa con respecto a la supervivencia general entre los tratamientos (RRI: 0.82; P = 0.29).
En las pacientes con afectación ganglionar, FEMARA® redujo de forma significativa el riesgo de mortalidad en 40% (RRI: 0.61; P = 0.035), pero no se observó ninguna diferencia significativa en las pacientes sin afectación ganglionar (RRI: 1.36; P = 0.385), ni tampoco en las pacientes con quimioterapia previa o sin quimioterapia. Los resultados se resumen en las tablas 4 y 5.
En materia de seguridad y de eficacia, no hubo ninguna diferencia entre las pacientes menores de 65 años y las mayores de esa edad.
Los acontecimientos adversos siguientes con independencia de su causalidad, se registraron con significativa mayor frecuencia en el grupo de FEMARA® que en el del placebo: rubefacción o sofocos (49.7% frente a 43.3%), artralgia/artritis (27.7% frente a 22.2%) y mialgia (9.5% frente a 6.7%). La mayoría de estos acontecimientos adversos se observaron durante el primer año de tratamiento. La incidencia de signos de osteoporosis notificados por la paciente fue mayor en el grupo de FEMARA® que en el grupo del placebo (6.9% frente a 5.5%). La incidencia de fracturas clínicas apenas fue más elevada en las pacientes que recibieron FEMARA® que en las que recibieron el placebo (5.9% frente a 5.5%). La tasa de fracturas por 1,000-mujeres-año en el grupo de letrozol (24.6) se ubica dentro del intervalo de valores que se observan en mujeres sanas posmenopáusicas de la misma edad.
Los primeros resultados del subestudio de densidad mineral ósea (el seguimiento duró una mediana de 20 meses) indican que al cabo de dos años y en comparación con los datos iniciales, las pacientes que recibieron letrozol experimentaron una reducción media de 3% de la densidad mineral ósea de la cadera en comparación con el 0.4% del grupo placebo (P = 0.048). No hubo ninguna diferencia significativa con respecto a los cambios de la densidad mineral ósea de la columna lumbar. Los primeros resultados del subestudio de lípidos (el seguimiento duró una mediana de 29 meses) no revelaron ninguna diferencia significativa entre los grupos de FEMARA® y del placebo. En el estudio de base (core study), la incidencia de acontecimientos cardiovasculares isquémicos fue comparable entre los grupos de tratamiento (6.8% frente a 6.5%).
Tratamiento de primera línea: Se llevó a cabo un ensayo doble-ciego debidamente controlado, en el que se compararon los efectos de 2.5 mg de letrozol (FEMARA®) con los del tamoxifeno como tratamiento de primera línea en mujeres posmenopáusicas con cáncer de mama local avanzado o metastásico. En 907 mujeres, el letrozol (FEMARA®) fue superior al tamoxifeno respecto al tiempo transcurrido hasta la progresión del tumor (criterio de valoración principal), remisión objetiva general del tumor, tiempo transcurrido hasta el fracaso terapéutico y beneficio clínico. Los resultados correspondientes se presentan en la tabla 6.
Tanto el tiempo tanscurrido hasta la progresión del tumor como el porcentaje de remisión objetiva fueron significativamente más largos o mayores con FEMARA® que con el tamoxifeno, con independencia de la presencia de receptor (tabla 7).
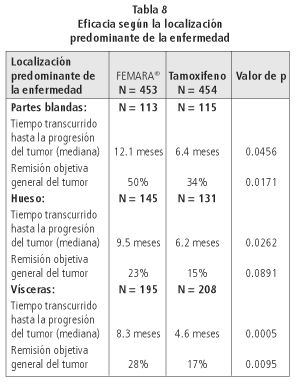
La eficacia según la localización predominante de la enfermedad se describe en la tabla 8.
>
El diseño del estudio permitió a las pacientes cambiar de tratamiento, en caso de progresión tumoral, o bien, abandonar el estudio. Cerca del 50% de pacientes pasó al grupo terapéutico opuesto y el pase fue prácticamente completo a los 36 meses. La mediana de tiempo transcurrido hasta el cambio de tratamiento fue de 17 meses (de FEMARA® a tamoxifeno) y de 13 meses (de tamoxifeno a FEMARA®). Como tratamiento de primera línea en pacientes con cáncer de mama avanzado, FEMARA® se asocia a una mayor supervivencia en comparación con el tamoxifeno.
La mediana de supervivencia fue de 34 meses con FEMARA® y de 30 meses con tamoxifeno.
La comparación de las cifras de FEMARA® y del tamoxifeno revela que un número significativamente mayor de pacientes que recibieron FEMARA® seguían vivas durante los primeros 24 meses del estudio (según la prueba log-posicional repetida), véase tabla 9.
Los efectos del tratamiento analizados a través de la covariable terapia antiestrogénica adyuvante previa se detallan en la tabla 10.
En las pacientes que no pasaron al grupo terapéutico opuesto, la mediana de supervivencia fue de 35 meses con FEMARA® (N = 219 IC del 95%: 29 a 43 meses) frente a 20 meses con tamoxifeno (N = 229, IC del 95%: 16 a 26 meses).
La duración total de tratamiento endocrino (tiempo transcurrido hasta la quimioterapia) fue significativamente mayor con FEMARA® (mediana de 16.3 meses, IC de 95%: 15 a 18 meses) que con el tamoxifeno (mediana de 9.3 meses, IC del 95%: 8 a 12 meses) (prueba log-posicional p = 0.0047).
Se observó un deterioro de la puntuación de Karnofsky de 20 puntos o más en un número significativamente menor de pacientes que recibieron letrozol como tratamiento de primera línea (19%) que en las pacientes que recibieron el tamoxifeno como tratamiento de primera línea (25%) (razón de ventajas, p = 0.0208).
Tratamiento de segunda línea: Se llevaron a cabo dos ensayos clínicos debidamente controlados en los que se compararon dos dosis de letrozol (FEMARA® 0.5 y 2.5 mg) con el acetato de megestrol y la aminoglutetimida, respectivamente, en mujeres posmenopáusicas con cáncer de mama avanzado que habían sido tratadas previamente con antiestrógenos.
Al comparar la dosis de 2.5 mg de letrozol (FEMARA®) con el acetato de megestrol, se apreciaron diferencias estadísticamente significativas en favor del primero en el porcentaje de respuesta objetiva general del tumor (24% frente a 16% p = 0.04) y en el tiempo transcurrido hasta que se observaba un fracaso terapéutico (p = 0.04). El tiempo transcurrido hasta observar progresión no fue significativamente distinto entre la dosis de 2.5 mg de letrozol (FEMARA®) y el acetato de megestrol (p = 0.07). No hubo ninguna diferencia de supervivencia general estadísticamente significativa entre ambos grupos terapéuticos (p = 0.2).
En el segundo estudio, la dosis de 2.5 mg de letrozol fue estadísticamente superior a la aminoglutetimida en lo que respecta al tiempo transcurrido hasta la progresión del tumor (p = 0.008), el tiempo transcurrido hasta el fracaso terapéutico (p = 0.003) y la supervivencia general (p = 0.002). No hubo ninguna diferencia significativa en la tasa de remisión entre la dosis de 2.5 mg de letrozol y la aminoglutetimida (p = 0.06).
Tratamiento prequirúrgico: Se llevó a cabo un estudio doble-ciego en 337 pacientes distribuidas al azar en el grupo de 2.5 mg de letrozol (FEMARA®) o del tamoxifeno durante cuatro meses. La evaluación clínica indicó 55% de remisión objetiva del tumor en el grupo de pacientes tratadas con FEMARA® y 36% de remisión objetiva del tumor en el grupo que recibió el tamoxifeno (p < 0.001).
Este resultado fue confirmado constantemente por medio de ecografía (p = 0.042) y mamografía (p < 0.001), que proporcionaron la estimación más comedida de remisión.
Esta remisión redundó en un número estadísticamente mayor de pacientes en el grupo de FEMARA® que se volvieron aptas para recibir (y que recibieron) un tratamiento de conservación de mamas (45% de pacientes del grupo de FEMARA® frente al 35% del grupo de tamoxifeno; p = 0.022).