Farmacodinamia:
Mecanismo de acción: La enfuvirtida es el primer miembro del grupo terapéutico denominado inhibidores de la fusión.
Se trata de un inhibidor de la reordenación estructural de la gp41 del VIH-1, que actúa uniéndose específicamente a esta proteína del virus fuera de las células y bloqueando así la entrada del virus en ellas.
La enfuvirtida no precisa activación intracelular. La actividad antiviral de la enfuvirtida se debe a su asociación con la heptada repetida HR1, situada dentro de la gp41 nativa de la superficie vírica.
Actividad antiviral in vitro: La actividad antiviral in vitro de la enfuvirtida ha quedado demostrada en la infección aguda de líneas celulares linfoblastoides T, células de la serie monocítica/macrofágica y células mononucleares primarias de sangre periférica (PBMC) por cepas de VIH-1 de laboratorios y clínicas.
La enfuvirtida demostró actividad selectiva anti-VIH-1 frente a cepas prototípicas y primarias del virus. La sensibilidad de 130 cepas víricas basales de PBMC a la enfuvirtida se determinó en un análisis de células cMAGI de la CE50 de la enfuvirtida frente a estas cepas víricas fue de 0.016 µg/ml (DE = 0.057), con un intervalo de valores de < 0.001 a 0.480 µg/ml.
La enfuvirtida también inhibía la fusión intercelular mediada por la cubierta del VIH-1. Los estudios de uso combinado de la enfuvirtida con miembros representativos de los distintos grupos de antirretrovirales (inhibidores nucleosídicos de la transcriptasa inversa, inhibidores no nucleosídicos de la trascriptasa inversa e inhibidores de la proteasa; a saber: con zidovudina, lamivudina, nelfinavir, indinavir y efavirenz) revelaron efectos aditivos o sinérgicos y la ausencia de antagonismo. No se ha establecido ninguna relación entre la sensibilidad in vitro del VIH-1 a la enfuvirtida y la inhibición de la replicación de VIH-1 en el ser humano.
Dado que los blancos enzimáticos son diferentes, y como se desprende de la actividad de la enfuvirtida contra las otras cepas de VIH resistentes a la enfuvirtida deberían permanecer sensibles a los inhibidores nucleosídicos de la transcriptasa inversa, los inhibidores no nucleosídicos de la transcriptasa inversa y los inhibidores de la proteasa.
Resistencia in vitro: Se han seleccionado in vitro cepas de VIH-1 con menor sensibilidad a la enfuvirtida que contienen sustituciones en los aminoácidos 36-38 del ectodominio de la gp41.
Estos cambios se correlacionan con grados variables de disminución de la sensibilidad a la enfuvirtida de cepas mutantes de VIH diseñadas por mutagénesis dirigida.
Resistencia in vivo: En la aparición de resistencia a la enfuvirtida influye la eficacia de todo el régimen terapéutico. Sustituciones de los aminoácidos 36-45 de gp41 durante el tratamiento se han observado virus de pacientes tratados con FUZEON® en los estudios clínicos de fases II y III. Las sustituciones de aminoácidos observadas, en orden establecido, no existe ninguna relación entre estas sustituciones y la eficacia in vivo del tratamiento.
Los cambios en los aminoácidos 36-45 de gp41 durante el tratamiento suelen entrañar una menor sensibilidad fenotípica in vitro de las cepas víricas de estos pacientes a la enfuvirtida.
Resistencia cruzada: La enfuvirtida tiene igual eficacia in vitro a cepas de laboratorio y clínicas en estado natural (salvajes), así como frente a las que presentan resistencia a 0, 1, 2 ó 3 grupos de antirretrovirales (inhibidores nucleosídos de la transcriptasa inversa, inhibidores no nucleosídicos de la transcriptasa inversa e inhibidores de la proteasa).
Estas cepas poseían resistencia genotípica identificada específicamente frente a zidovudina, lamivudina, estavudina, didanosina, zalcitabina, abacavir, nevirapina, delavirdina, efavirenz, indinavir, saquinavir, nelfinavir, ritonavir y amprenavir; todas ellas eran sensibles a la enfuvirtida. Inversamente, las mutaciones en los aminoácidos 36-45 de gp41, que confieren resistencia a la enfuvirtida, no deberían ocasionar resistencia cruzada a otros grupos de antirretrovirales.
Eficacia (estudios clínicos):
Estudios en pacientes tratados anteriormente con antirretrovirales: En los estudios T20-301 (TORO-1) y T20-302 (TORO-2) en marcha, de diseño aleatorizado, controlado, abierto y multicéntrico participan pacientes infectados por el VIH-1 y tratados anteriormente con inhibidores nucleosídicos y no nucleosídicos de la transcriptasa inversa e inhibidores de la proteasa, o bien con intolerancia o resistencia documentada a estos fármacos.
Todos los pacientes recibieron un tratamiento optimizado (TO), compuesto por 3 a 5 antirretrovirales, seleccionados de acuerdo con los tratamientos previos del paciente y con los análisis iniciales de la resistencia vírica genotípica y fenotípica. La distribución de los pacientes a los grupos de tratamiento con fuzeon® (90 mg dos veces al día) + TO o TO sólo fue aleatorizada, a razón de 2:1.
Estudios TORO-1: FUZEON® + tratamiento optimizado frente a tratamiento optimizado solamente.
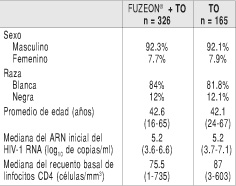
En el estudio TORO-1, la población con intención de tratar era de 491 pacientes, cuyos datos demográficos se recogen en la tabla 1. Los pacientes habían recibido anteriormente 12 antirretrovirales (mediana) durante 7 años (mediana).
Estudio TORO-2: FUZEON® + tratamiento optimizado frente a tratamiento optimizado solamente.
En el estudio TORO-2, la población con intención de tratar era de 504 pacientes, cuyos datos demográficos se recogen en la tabla 2.
Los pacientes habían recibido anteriormente 12 antirretrovirales (mediana) durante 7 años (mediana) .
Tabla 2. Datos demográficos de los pacientes del estudio Toro -2
La tabla 3 resume los cambios registrados en ambos estudios, al cabo de 24 semanas, en los valores plasmáticos de ARN del VIH-1 (log10 de copias/ml), el recuento de linfocitos CD4, el porcentaje de los pacientes con disminución del ARN del VIH ³ 1 log con respecto a los valores basales, pacientes con < 400 copias/ml y pacientes con < 50 copias/ml; todos estos cambios fueron significativamente mayores en el grupo tratado con FUZEON®.
+ ITT: pacientes que recibieron tratamiento y se sometieron a un análisis de control del ARN después del basal.
* Valor estadísticamente significativo: p < 0.05.
Media de mínimos cuadrados (LOCF). Promedio de los dos últimos valores.
§ Retirada del tratamiento y fracaso virológico = fracaso. Dos visitas consecutivas para confirmar la respuesta virológica.
Niños: Los datos disponibles sobre la eficacia de FUZEON® en niños mayores de 3 años son limitados.
El estudio T20-204, en marcha, es un ensayo clínico abierto y multicéntrico para evaluar la farmacocinética, la seguridad toxicológica y la actividad antiviral de FUZEON® en 14 niños de 3 a 12 años, tratados ya con al menos 2 grupos de antirretrovirales aprobados.
En el estudio T20-204, al régimen antirretroviral de fondo ya existente se agregó una dosis de 30 ó 60 mg/m2 de FUZEON® dos veces al día.
Al cabo de 7 días, el régimen de fondo se cambió a 3 nuevos o sensibles antirretrovirales y la dosis de FUZEON® se mantuvo. La mediana de edad de los pacientes era de 8 años (extremos: 3.7 y 11.9 años).
La mediana basal de linfocitos CD4 era de 523/µl, la mediana basal de copias de ARN del VIH por ml era de 4.8 log. Tras el análisis de seguridad, farmacocinética y actividad antiviral después de 7 días, todos los pacientes salvo uno recibieron una dosis de 60 mg/m² de FUZEON®. La mediana de la diferencia del número de copias de ARN del VIH por ml en el día 7o. con respecto a la cifra basal fue de 1.15 log10 en 9 niños tratados con la dosis de 60 mg/m².
Salvo 3 pacientes, todos los demás terminaron las 48 semanas de tratamiento. En la semana 48a, 6 de 14 (43%) pacientes presentaban una disminución > 1 logro del número de copias de ARN del HIV-1 y 4 de 14 (29%) pacientes se encontraron por debajo de 400 copias/ml. La mediana de las variaciones con respecto a los valores basales de copias de ARN del VIH-1 por ml y de número de linfocitos CD4 era, respectivamente, de 1.24 log10 y 237 células/µl.
Farmacocinética: Las propiedades farmacocinéticas de la enfuvirtida se han investigado en adultos y niños infectados por el VIH-1.
Absorción: Tras la inyección subcutánea en el abdomen de una dosis única de 90 mg de FUZEON® a 12 pacientes infectados por el VIH-1, la Cmáx media (± DE) era de 4.59 ± 1.5 µg/ml; el área bajo la curva de concentraciones plasmáticas (ABC), de 55.8 ± 12.1 µg/ml, y la biodisponibilidad absoluta (utilizando la dosis I.V. de 90 mg como referencia), del 84.3% ± 15.5%.
La absorción subcutánea de la enfuvirtida es proporcional a la dosis administrada en el intervalo de 45 a 180 mg. La absorción subcutánea de la dosis de 90 mg es comparable cuando se inyecta en el abdomen, el muslo o el brazo.
La figura 1 muestra la concentración plasmática media en estado de equilibrio de la enfuvirtida en dosis de 90 mg.
Figura 1. Concentración plasmática en estado de equilibrio de la enfuvirtida* en dosis de 90 mg dos veces al día, n = 11
* Barra de error = desviación estándar.
En cuatro estudios distintos (n = 9.12), el promedio de la concentración plasmática mínima en estado de equilibrio se situó entre 2.6 y 3.4 µg/ml.
Distribución: El volumen medio (± DE) de distribución en equilibrio tras la administración intravenosa de una dosis de 90 mg de FUZEON® (n = 12) era de 5.5 ± 1.1 l. La enfuvirtida se une en un 92% a las proteínas del plasma infectado por el VIH, en un intervalo de concentraciones plasmáticas de 2 a 10 µg/ml. La enfuvirtida se une sobre todo a la albúmina y, en menor medida, a la glucoproteína ácida a-1. El saquinavir, nelfinavir, lopinavir, efavirenz, nevirapina, amprenavir, itraconazol, midazolam y warfarina, no desplazan a la enfuvirtida de sus sitios de unión. Por otro lado, la enfuvirtida tampoco desplaza de sus sitios de unión al efavirenz, amprenavir, midazolam ni la warfarina.
Metabolismo: La enfuvirtida siendo un péptido se cataboliza en sus aminoácidos constituyentes, los cuales, a continuación, se reciclan dentro del organismo.
Los estudios in vitro con microsomas humanos indican que la enfuvirtida no inhibe las enzimas del citocromo P-450.
En estudios in vitro con microsomas y hepatocitos humanos, la hidrólisis del grupo amida de la fenilalanina carboxiterminal da lugar a un metabolito desamidado, cuya formación no depende del NADPH. Este metabolito se detecta en el plasma humano después de administrar la enfuvirtida, con un valor de ABC que varía entre el 2.4 y el 15% del ABC de la enfuvirtida.
Eliminación: No se han efectuado estudios de balance de masas para determinar la(s) vía(s) de eliminación de la enfuvirtida en el humano. Ahora bien, los estudios con roedores tratados con enfuvirtida radiomarcada con H3 mostraban una recuperación incompleta de la radiactividad en los excrementos en los animales al cabo de 7 días de la administración y retención de radiactividad en los músculos esqueléticos.
Tras una dosis subcutánea de enfuvirtida de 90 mg (n = 12), la semivida media de eliminación (± DE) es de 3.8 ± 0.6 h, y el aclaramiento medio (± DE), de 1.7 ± 0.4 l/h.
Farmacocinética en poblaciones especiales:
Insuficiencia hepática: No se ha estudiado la farmacocinética de la enfuvirtida en pacientes con insuficiencia hepática.
Insuficiencia renal: No se ha estudiado la farmacocinética de enfuvirtida en pacientes con insuficiencia renal. Sin embargo, el análisis de los datos de la concentración plasmática en los pacientes de los ensayos clínicos revela que el aclaramiento de la enfuvirtida no se ve afectado cuando el aclaramiento de creatinina es mayor de 35 ml/min.
Sexo y peso: El análisis de los datos de la concentración plasmática en los pacientes de los ensayos clínicos puso de relieve que el aclaramiento de la enfuvirtida es un 20% menor en el sexo femenino que en el masculino, y que aumenta con el peso corporal independientemente del sexo (20% superior en los pacientes de 100 kg y 20% inferior en los de 40 kg, en relación con un paciente prototipo de 70 kg). Ahora bien, esas variaciones carecen de importancia clínica y no se precisa ningún ajuste posológico.
Raza: El análisis de los datos de la concentración plasmática en los pacientes de los ensayos clínicos indica que el aclaramiento de la enfuvirtida no difiere entre las personas de raza negra y de raza blanca. Otros estudios farmacocinéticos tampoco muetran diferencias entre los asiáticos y los blancos una vez ajustada la exposición en función del peso corporal.
Ancianos: No se ha estudiado la farmacocinética de la enfuvirtida en pacientes mayores de 65 años.
Niños: Se ha estudiado la farmacocinética de la enfuvirtida en 32 niños y adolescentes de 3 a 16 años, tratados con dosis de 0.5 a 2.5 mg/kg. La dosis de 2 mg/kg dos veces al día (máximo de 90 mg dos veces al día) produjo concentraciones plasmáticas de enfuvirtida similares a las obtenidas en adultos tratados con 90 mg dos veces al día.
En 20 niños y adolescentes de 5 a 16 años tratados con una dosis de 2 mg/kg dos veces al día se obtuvieron los valores siguientes: ABC medio en equilibrio estable, 51.4 ± 22.8 µg/h/ml; Cmáx, 5.81 ± 2.35 µg/ml, y Cmáx, 2.82 ± 1.46 µg7/ml.